肯尼-卡菲综合征



临床病史
一名17岁女性,患有匀称性侏儒症和先天性甲状旁腺功能减退症。
影像学表现
一名17岁女性患者,既往有先天性甲状旁腺功能减退和手足抽搐病史。自童年起即受严重侏儒症影响,缺乏青春期生长突增。临床检查所见为匀称侏儒症(身高120厘米,体重31公斤);特征性面容包括前额宽大和局部脱发区域、三角面、眼球不对称及小眼球、下颌不对称并伴有牙齿发育异常,腰椎侧弯及膝内翻。
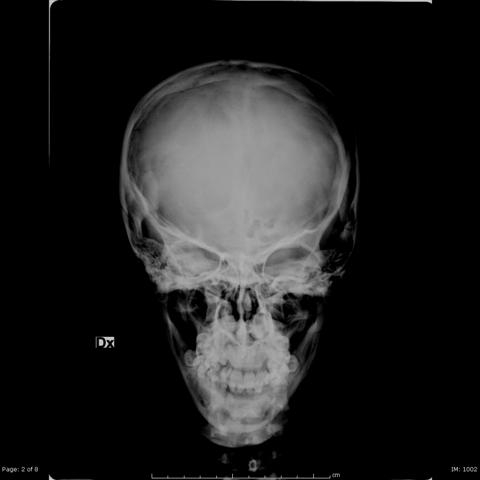
骨骼X线片检查显示头颅(图1)及长骨皮质增厚,同时髓腔变窄(图2)。
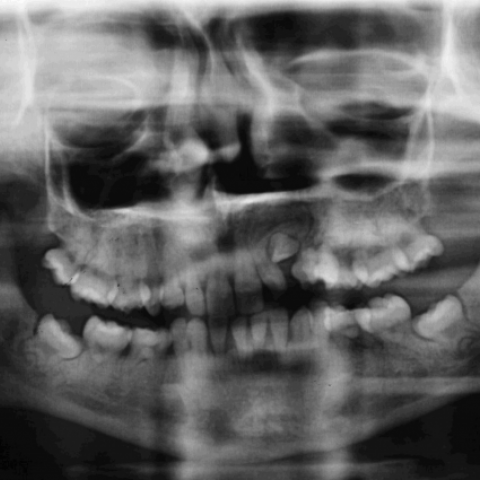
全景牙片检查还显示多颗牙胚和恒牙缺失,并存在牙冠发育不良及扁平(图3)。
基因检测提示22q11微缺失,高度提示Kenny-Caffey综合征。
病情讨论
Kenny-Caffey综合征是一种罕见的综合征,其特点包括生长迟缓、长骨均匀细长且伴有骨髓腔狭窄、骨皮质增厚、低钙血症(可能在早期出现手足痉挛)、高磷血症、眼部异常以及智力正常。
这些儿童具有典型面容:眼窝深陷、鼻梁塌陷并带有鹰钩鼻、人中加长、上唇薄、小下颌、大而柔软的耳垂、头围增大、额部隆起、前囟门闭合延迟、明显的额缝增宽以及颅内无板障空间。眼部改变包括小眼球症、远视、假性视乳头水肿、血管迂曲、黄斑区混浊、角膜和视网膜钙化,以及眉毛和睫毛稀疏。也可能出现龋齿、掌指骨短缩以及发际线高。偶尔有耳聋的报道。所有患儿的主要症状表现为在出生后数天或数周内出现的低钙血症性手足搐搦或全身抽搐。偶尔症状可延至生后第四至第七个月才出现。喂养障碍、呕吐和腹泻是其他常见问题。综合征中可能出现的其他表现包括指甲发育不良及新生儿肝病。另外也有报告显示存在小睾丸(microorchidism)。
Kenny-Caffey综合征的实验室缺陷包括血清钙和镁水平低、血清磷水平高、新生儿甲状旁腺功能减退,有时还伴有贫血、嗜酸性粒细胞增多和持续性中性粒细胞减少。该综合征的免疫学缺陷特点是存在特异性T细胞异常,即抑制性T细胞(CD8)比例升高从而导致辅助/抑制T细胞比率降低,以及对有丝分裂原刺激的T细胞反应降低。
放射学检查显示大多数患儿存在骨髓腔狭窄、骨致密化(osteosclerosis)及长骨皮质增厚。再结合他们的低钙血症、高磷血症以及部分患儿中免疫反应性甲状旁腺激素浓度低的情况,这些发现支持了该诊断。
鉴别诊断包括其他干骺端发育不良。在Camurati-Englemann病中,特征是长骨和颅底的对称性骨质增生,常可见进行性听力损害(如进行性耳聋和耳痛)、肌病和神经系统功能障碍。Hardcastle综合征的放射学表现为骨髓腔狭窄、骨皮质增厚以及骨梗死区。临床表现包括慢性疼痛(可发展至丧失活动能力)、潜在的病理性骨折(可导致延迟愈合或不愈合)以及恶性肿瘤风险。迟发型骨硬化症(Osteopetrosis tarda)会导致所有骨骼密度增加,伴有骨皮质增宽和骨髓腔变窄。然而此病少有症状,可能在青少年或成年后仅因病理性骨折而被发现。先天性骨硬化症(Osteopetrosis congenita)在出生时即存在并导致严重的残障,骨骼对骨髓的侵占可导致全血细胞减少、溶血、贫血及肝脾肿大,经常因反复出血或感染而在幼年早期死亡。
在Kenny-Caffey综合征中,已有文献报道存在22q11.2单体不足,这使CATCH 22微缺失的范围扩大,以涵盖Kenny-Caffey综合征。而本患儿的核型分析则显示正常。
长期补充钙和维生素D被推荐用于治疗。预后可能存在差异。Kenny-Caffey综合征的主要结局是身材矮小,精神发育能力很少受到影响。有文献报道死亡率为33%,原因是精神运动发育迟缓和并发感染。
鉴别诊断列表
最终诊断
肯尼-卡菲综合征 (KCS)。
证书
(无)
图像分析
颅骨X线片

骨骼X线检查




曲面断层全景X线摄影
影像学发现
1. 颅骨:从正位与侧位颅骨X线片可观察到患者颅盖骨呈增大趋势,颅顶结构增厚,颅板内外板间的双板层空间(diploic space)减小甚至显示不明显,前额部分有一定程度的突出(额隆突)。
2. 四肢长骨:下肢(胫腓骨)及上肢(肱骨、尺桡骨)X线示骨质紧密且骨皮质增厚,骨髓腔狭窄或呈梗阻性改变。尤其是骨干部位可见髓腔变窄,整体呈“细长且致密”外观。
3. 骨盆及髋部:髂骨形态无明显结构异常,但同样可见皮质增厚趋势,骨小梁排列较致密。
4. 牙齿/牙槽骨:全景片提示牙齿发育大致正常,但可见牙周骨质密度增加,局部骨髓腔表现相对纤细,尚未见明显牙齿缺失或大的病变。
总体而言,影像特点符合长骨骨皮质增厚、骨髓腔狭窄、颅盖骨增厚与前额突出等表现。
潜在诊断
-
Kenny-Caffey综合征(首要考虑)
结合患者存在的先天性甲状旁腺功能减退、身材矮小(和谐性身材发育不良)、高磷血症及低钙血症病史,影像学上出现长骨骨髓腔狭窄并伴骨质致密增厚,符合Kenny-Caffey综合征的典型表现。 -
Camurati-Engelmann病
该病常表现为对称性骨皮质增厚、骨干硬化及颅底增厚,可引起骨痛、肌无力与神经症状,但通常与低钙血症、先天性甲状旁腺功能减退并无明确关联,故在本例中可能性较低。 -
Hardcastle综合征
也有骨皮质增厚与髓腔变窄表现,但常有骨梗死、慢性疼痛、病理性骨折、骨不连等,更倾向于此类特征性病变,本例尚不完全吻合。 -
骨硬化症(Osteopetrosis)
呈骨质致密增多,骨髓腔狭窄或消失,但本病常伴发早期贫血、脾肿大等造血功能问题,且临床罕见先天性甲状旁腺功能减退。故与本例临床表现不完全一致。
最终诊断
结合患者17岁女性、先天性甲状旁腺功能减退、低血钙与高血磷、身材矮小及X线所见的长骨髓腔狭窄、骨皮质增厚,最符合 Kenny-Caffey综合征 的诊断。 若需更进一步确认,可考虑检测血清甲状旁腺激素(PTH)水平、基因检测及免疫学评估等,以最终明确诊断。
治疗方案与康复计划
1. 药物及补充治疗:
- 长期补充钙剂与维生素D(含活性维生素D制剂)以维持正常血钙水平,预防低钙惊厥及骨质异常变化。
- 若合并低镁,可适当补充镁剂,以协同维持血钙平衡。
- 定期监测血液钙、磷、镁及PTH水平,必要时调整剂量。
2. 其他支持治疗:
- 根据免疫学评估结果,若有免疫减弱,可需做适当免疫功能监测,必要时由专科医生评估干预。
- 保持营养均衡,充分摄入蛋白质、维生素及微量元素,以促进生长发育。
3. 康复与运动处方:
鉴于患者骨质形态特别,需在身体条件允许的前提下逐步进行运动训练,以提升骨骼肌肉系统的功能,预防骨量流失。以下为简单可执行的原则:
- 频率(Frequency):建议每周3~4次运动,避免过度疲劳。
- 强度(Intensity):初始以低至中等强度为主,选择不产生剧烈震荡或过大负荷的项目,如步行、游泳、骑自行车等。
- 时间(Time):每次持续20~30分钟,视身体情况可逐渐延长。
- 方式(Type):
1) 关节保护型有氧运动:如在支撑条件下的固定自行车或游泳,能减少对下肢骨骼的冲击。
2) 轻量抗阻练习:可使用弹力带或极轻的哑铃,以训练上肢及躯干肌力,初期每组10~12次,2~3组为宜。
- 进阶(Progression):当身体适应后,可小幅增加运动强度(如略增步行距离或速度),或稍增抗阻训练的负重,但应注意监测血 calcium 水平及身体耐受度。
在运动过程中,应警惕低钙抽搐或疲劳骨折的风险,定期复查骨密度并观察血生化指标,必要时在专科康复科或骨科指导下完善康复计划。
免责声明
本报告仅为基于现有影像和临床资料所做的参考性分析,不能替代线下面诊或专业医生的综合评估和诊治。若有任何疑问或病情变化,请及时就医或咨询专科医生。
人类医生最终诊断
肯尼-卡菲综合征 (KCS)。