Kenny-Caffey Syndrome.



Clinical History
A 17-year-old female with harmonic dwarfism and congenital hypoparathyroidism.
Imaging Findings
A 17-year-old female patient presented with a history of congenital hypoparathyroidism and carpo-podalic convulsions. Since childhood she was affected by severe dwarfism with lack of puberal growth spurt. The clinical examination described harmonic dwarfism (height of 120 cm with a weight of 31 kg); a characteristic facies with large forehead and alopecia areas, triangular face, asymmetric ocular bulbs and nanophtalmic eyes, asymmetric jaw with dentition anomalies, lumbar scoliosis and genu varum.
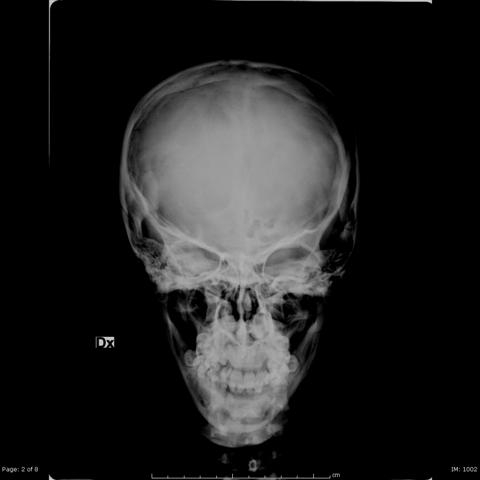
Radiographies of the skeleton were performed and showed cortical thickening of cranium (Fig. 1) and long bones, with reduction of medullary cavity (Fig. 2).
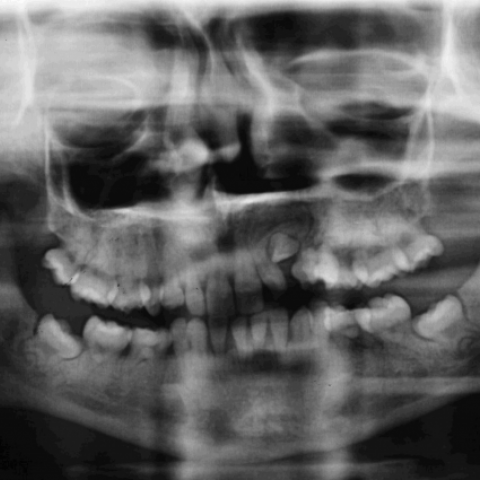
Orthopantomography was also performed and showed lack of numerous tooth buds and permanent teeth with hypoplasia and flattening of the crowns (Fig. 3).
Genetic test reported a microdeletion on 22q11, strongly suggestive of Kenny-Caffey syndrome.
Discussion
Kenny-Caffey Syndrome is a rare syndrome characterised by growth retardation, uniformly small slender long bones with medullary stenosis, thickened cortex of the long bones, hypocalcemia possibly with tetany at an early age, hyperphosphatemia, ocular abnormalities, and normal intelligence.
The children have characteristic facies with deep-set eyes, depressed nasal bridge with a beaked nose, long philtrum, thin upper lip, micrognathia, large floppy earlobes, macrocephaly, frontal bossing, delayed closure of anterior fontanel, wide metopic suture and absent diploic space in the skull. The changes in the eyes include microphthalmia, hyperopia, pseudopapilledema, vascular tortuosity, macular clouding, corneal and retinal calcifications and sparse eyebrows and eyelashes. There may be also dental caries, brachymetacarpalgia and high hairline. Occasional deafness has been reported. The presenting complaints in all children is hypocalcemic tetany or generalised convulsions detected in the first few days or weeks of life. Occasionally symptoms may be delayed to fourth to seventh month of life. Feeding disorders, vomiting, and diarrhoea were other common problems encountered. Other traits with variable presentation in the syndrome are hypoplasic nails and neonatal liver disease. The presence of microorchidism has also been reported.
Laboratory defects in Kenny-Caffey syndrome include the presence of low serum calcium and magnesium, high serum phosphorus, neonatal hypoparathyroidism, sometimes with anaemia, eosinophilia, and persistent neutropenia. The immunological defect in Kenny-Caffey syndrome is characterised by the presence of a specific T cell abnormality in terms of increased suppressor fraction CD8 with a consequent low helper/suppressor ratio, in addition to reduced T cell response to mitogens.
The radiological evidence of medullary stenosis, osteosclerosis and cortical thickening of long bones was found in most of them. This together with the hypocalcemia, hyperphosphatemia and low concentration of immunoreactive parathyroid hormone in some of them supports the diagnosis.
Differential diagnosis includes other diaphyseal dysplasias. Camurati-Englemann disease is characterized by symmetrical hyperostosis of the long bones and the skull base, often with progressive audiological signs such as progressive deafness and otalgia, myopathies and neurological disturbances.6 In Hardcastle syndrome radiological findings demonstrate medullary stenosis, cortical bone thickening, and areas of bone infarction. Clinical manifestations include chronic pain that can become incapacitating, potential pathological fractures with subsequent delayed or nonunion, and risk of malignancy. Osteopetrosis tarda leads to increased density of all bones, with widened cortices and narrowed medullary canals. However, this condition rarely causes symptoms and may be discovered in adolescents or adulthood after a pathological fracture. Osteopetrosis congenita is present at birth and causes severe disability. Bone encroachment on marrow results in pancytopenia, hemolysis, anemia, and hepatosplenomegaly. Repeated hemorrhage or infection usually leads to death in early childhood.
The presence of 22q11.2 haploinsufficiency was reported in Kenny-Caffey syndrome, which widens the spectrum of CATCH 22 microdeletion to accommodate Kenny-Caffey syndrome. Karyotyping in this child was normal.
Long-term treatment with calcium and vitamin D supplements is recommended. Prognosis may be variable. The primary outcome of Kenny-Caffey syndrome is short stature. Mental abilities are rarely affected. Mortality of 33% has been reported, because of psychomotor delay and intercurrent infections.
Differential Diagnosis List
Final Diagnosis
Kenny-Caffey Syndrome (KCS).
Liscense
Figures
Skull Radiographies

Skeleton Radiographies




Orthopantomography
Imaging Findings
1. Skull: Based on the frontal and lateral skull X-rays, the patient’s calvarium appears enlarged. The vault shows thickened structures, and the diploic space between the inner and outer tables is narrowed or even not clearly visible. There is some degree of frontal bossing.
2. Long Bones of the Extremities: X-rays of the lower limbs (tibia and fibula) and upper limbs (humerus, radius, and ulna) show increased bone density with thickened cortical bone and a narrowed or obstructed medullary cavity. Particularly in the diaphyseal region, the marrow canal is narrowed, giving an overall “elongated and dense” appearance.
3. Pelvis and Hip: The iliac bones do not show significant structural abnormalities, but similarly exhibit cortical thickening and densely arranged trabeculae.
4. Teeth/Alveolar Bone: The panoramic radiograph indicates generally normal tooth development, but with increased alveolar bone density and a relatively slender marrow cavity in certain areas. No obvious tooth loss or large lesions are seen.
Overall, the imaging features are consistent with thickened cortical bone of the long bones, narrowed medullary cavities, a thickened cranial vault, and frontal prominence.
Potential Diagnoses
-
Kenny-Caffey Syndrome (Primary Consideration)
Given the patient’s congenital hypoparathyroidism, short stature (harmonious developmental delay), hyperphosphatemia, hypocalcemia, and imaging findings of narrowed long bone marrow cavities with increased bone density, these are classic presentations of Kenny-Caffey Syndrome. -
Camurati-Engelmann Disease
This disease typically presents with symmetrical cortical thickening, diaphyseal sclerosis, and thickening of the skull base, which may cause bone pain, muscle weakness, and neurological symptoms. However, it usually does not have a clear association with hypocalcemia or congenital hypoparathyroidism, making it less likely in this case. -
Hardcastle Syndrome
Also characterized by cortical thickening and narrowing of the marrow cavity, but often involves bone infarcts, chronic pain, pathological fractures, and nonunion, which are more characteristic than what is observed here. The current case does not fully match these features. -
Osteopetrosis
Shows generalized increased bone density and narrowing or obliteration of the marrow cavity. However, osteopetrosis commonly presents with early anemia, splenomegaly, and other hematopoietic issues, and congenital hypoparathyroidism is rare. Thus, it does not fully align with the clinical findings here.
Final Diagnosis
Considering the 17-year-old female patient with congenital hypoparathyroidism, hypocalcemia and hyperphosphatemia, short stature, and X-ray findings of narrowed medullary cavities and thickened cortical bone, the most consistent diagnosis is Kenny-Caffey Syndrome. If further confirmation is required, evaluations such as serum PTH levels, genetic testing, and immunological assessments can be considered for definitive diagnosis.
Treatment Plan and Rehabilitation
1. Medication and Supplementation:
- Long-term supplementation of calcium and vitamin D (including active vitamin D formulations) to maintain normal serum calcium levels and prevent hypocalcemic seizures and bone abnormalities.
- If accompanied by hypomagnesemia, appropriate magnesium supplementation may help maintain calcium balance.
- Regularly monitor serum calcium, phosphorus, magnesium, and PTH levels; adjust therapeutic dosages as needed.
2. Other Supportive Treatment:
- Depending on immunological assessments, if immune function is compromised, specialized monitoring or intervention may be needed under the supervision of a specialist.
- Maintain a balanced diet with adequate protein, vitamins, and trace elements to support growth and development.
3. Rehabilitation and Exercise Prescription:
Given the particular skeletal considerations, a gradual exercise program is recommended to enhance musculoskeletal function and prevent bone loss, guided by the following principles:
- Frequency: 3–4 sessions of exercise per week, avoiding excessive fatigue.
- Intensity: Begin with low to moderate intensity. Choose activities that minimize impact or overload, such as walking, swimming, or cycling.
- Time: Each session should last approximately 20–30 minutes, gradually increasing as tolerated.
- Type:
1) Joint-protective aerobic exercises: Such as stationary cycling or swimming, which help reduce stress on the lower limb bones.
2) Light resistance training: Using resistance bands or very light dumbbells to strengthen upper limb and trunk muscles. Start with 10–12 repetitions per set for 2–3 sets.
- Progression: As the patient adapts, the exercise intensity can be slightly increased (e.g., longer or brisker walks), or weights can be raised slightly. Monitor serum calcium levels and physical tolerance closely.
During exercise, be aware of the risk of hypocalcemic tetany or stress fractures. Regularly check bone density and blood chemistry. If necessary, consult a rehabilitation or orthopedic specialist to optimize the rehabilitation plan.
Disclaimer
This report is for reference only, based on current imaging and clinical data, and does not replace in-person medical consultation or professional evaluation and treatment. If you have any concerns or experience any changes in condition, please seek medical attention or consult a specialist promptly.
Human Doctor Final Diagnosis
Kenny-Caffey Syndrome (KCS).