丛状神经纤维瘤伴肢体肥大


临床病史
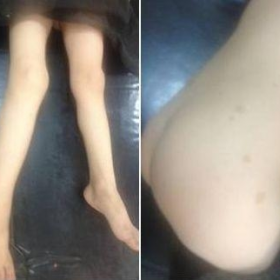
一名4岁男孩因右下肢肿块及双下肢不等长前来就诊。该肿块逐渐增大,导致间歇性搏动性疼痛。体格检查显示右小腿有一个质地坚实、可移动且触痛轻微的肿块。该男孩及其兄弟均有多枚色素沉着斑(Fig. 1)。
影像学表现
X线显示右腿较左腿更长。可见右侧胫骨和腓骨干轻度弯曲,并有软组织密度增加。(图2)
MRI显示右小腿后区有软组织肿块。该肿块呈浸润性,边缘分叶,自大腿远端及腘窝区一直延伸至足底。T1加权像上信号中等,T2加权像上信号高,加用钆剂后可见轻微强化。T2轴位图像可见肿块内多个靶征表现。(图3-4)
病情讨论
神经纤维瘤病是一种常染色体显性多系统遗传性疾病,由负责生成肿瘤抑制蛋白“神经纤维蛋白(neurofibromin)”的NF1基因发生缺失或突变引起。缺乏神经纤维蛋白会导致皮肤、骨骼和神经系统的畸形。临床确诊病例的患病率介于1/2000至1/5000之间。[1]
在NF1中骨骼常常受累:包括脊柱畸形、蝶骨和胫骨发育不良以及骨和软组织过度生长。[2]
在长骨发育不良中,最常见的部位是胫骨,约有7.1%的病例出现此情况。[2]
通常表现为胫骨成角、皮质变薄、骨折风险增大以及假关节形成。在此类病例中,肢体不等长通常是由受累肢体缩短引起的。然而,在没有发育不良的情况下也可能出现肢体过度生长,如本例中表现为较长的肢体伴随软组织肿块。有人推测,肿瘤相关性充血在某种程度上会诱发肢体过度生长。[3]
NF的诊断是基于临床。[4] 在本例中,通过符合七项临床标准中的三项(牛奶咖啡斑、胫骨发育不良以及一级亲属存在牛奶咖啡斑)确立诊断;这一临床背景及上述MRI表现提示为丛状神经纤维瘤(PN)的诊断。
PN是NF1的特征性病变,约有27%的病例出现PN。[5] 它是一种边界不清的浸润性肿块,累及神经、肌肉及邻近脂肪,因此通常无法实现完全切除。[6] PN常见于躯干和四肢,但也可累及头部、颈部、膀胱甚至肠系膜。[7]
在MRI上,PN表现为具有束状外观的浸润性肿块,在T1加权成像上信号偏低或与周围组织等信号,在T2加权成像上则表现为等信号或高信号,并且在钆增强后仅有轻微或无强化。一个典型的MR征象是轴位T2图像上的“靶征”:即胶原纤维构成的低信号中心,以及黏液样组织构成的高信号边缘。[8]
除临床表现外,MRI是诊断PN的主要手段。血管造影CT或MRI也有助于评估血管化情况,以便手术前栓塞。[9]
临床背景是诊断的最重要线索。当神经纤维瘤体积较大时,可导致受累肢体肥大,称为“神经性象皮肿”(elephantiasis neuromatosa)。[1]
鉴别诊断包括Proteus综合征,这是一种组织和骨骼的散发性疾病,特点是肢体过度生长、血管畸形和肌肉不对称[11, 12];以及Kasabach-Merritt综合征,其特征是血小板减少并伴有大范围浸润性软组织血管瘤。[13] 第一个综合征不太可能是本例诊断,因为本患者的疾病具有家族性;而第二种通常在T2上显示高信号并有明显强化,这与本例不符。
鉴别诊断列表
最终诊断
丛状神经纤维瘤
证书
没有可翻译的内容。
图像分析
躯干和下肢的照片

双腿X线检查

双腿核磁共振

双腿磁共振成像

1. 影像学发现
基于患儿的X线与MRI影像可见:
- 右侧小腿软组织肿块,边界较不清晰,呈浸润性生长,产生一定程度的骨性变化。X线提示右侧胫骨长度较对侧略长,且有骨质轻度弯曲。此与临床所见的患肢轻度过度生长相一致。
- MRI显示肿块在T1加权成像上呈低或等信号,在T2加权成像上呈高或混杂信号,局部可见“靶征”(中心相对低信号、周边高信号),提示内部存在胶原纤维和黏液样基质。
- 病变范围以神经束为中心,周围软组织亦受累,边缘较为不规则,符合典型的丛状神经纤维瘤影像学表现。
2. 潜在诊断
根据上述影像学表现以及患儿的临床病史(多块咖啡牛奶斑、家族史、局部肢体增粗畸形等),以下为可能考虑的诊断:
- 丛状神经纤维瘤(Plexiform neurofibroma):
- 在神经纤维瘤病I型(NF1)中常见,尤其是伴随咖啡牛奶斑等特征性体表表现。
- MRI典型可见“靶征”,呈浸润性生长,难以完整切除。
- 可导致局部肢体过度生长或“象皮肿样改变”。
- Proteus综合征:
- 表现为肢体及软组织过度生长,但多为散发,且无明显神经纤维瘤或家族史。
- 通常伴有骨骼畸形与其他系统异常,但本例中患儿有家族性发现,Proteus综合征可能性较低。
- 血管畸形或血管瘤(如Kasabach-Merritt综合征):
- 一般在成像上可见明显的血管走行异常,T2加权成像常高度增强,且可出现血小板减少等临床表现(Kasabach-Merritt)。
- 本例无显著血管高信号增强或血小板异常记录,故可能性偏低。
3. 最终诊断
综合患儿年龄、临床症状、家族史、多处咖啡牛奶斑、右小腿软组织逐渐增生及影像学典型表现,可明确诊断为神经纤维瘤病1型(NF1)合并丛状神经纤维瘤。
4. 治疗方案与康复计划
4.1 治疗策略
- 观察与定期随访:如肿瘤生长缓慢且症状轻微,可在专科医生指导下定期复查影像学和临床表现,监测病变大小与功能。
- 手术治疗:
- 若肿瘤进展明显、压迫神经血管造成功能障碍或出现持续性疼痛,可考虑手术部分切除或减压。
- 需注意丛状神经纤维瘤常呈弥漫性增殖,完整切除困难,术后仍有复发风险。
- 药物或基因治疗(研究阶段):目前对NF1相关神经纤维瘤的特异性药物治疗处于研究探索期,可根据最新进展和临床试验情况进行评估。
4.2 康复与运动处方
鉴于患儿的肢体发育尚在进行中,应在专科康复科医生的指导下进行个体化的运动与功能锻炼:
- 运动目标:在保护患肢的前提下,维持肌力与关节活动度,避免过度负重导致疼痛或病情加重。
- FITT-VP 原则:
- 频率(Frequency):每周2-3次轻度至中等强度活动。
- 强度(Intensity):控制心率与患肢承重程度,可从轻度(如低冲击力的游戏活动)开始。
- 时间(Time):每次运动时间约15-30分钟,视患儿耐受程度递增。
- 类型(Type):以关节及肌力维护为主,如轻度骑行、游泳(在专人陪护下)、简单平衡练习等。
- 进阶(Progression):根据肢体状况稳定性、X线和MRI复查结果,逐步增加活动时间和难度。
- 体量(Volume)与动机(Pattern):将运动融入患儿日常游戏,鼓励积极参与,避免疲劳和过度损伤。
- 安全注意事项:随时监测疼痛、关节形态变化以及肿瘤局部反应,如出现红肿、疼痛加剧,应及时就诊评估。
5. 免责声明
本报告为基于当前所提供信息的综合性医学分析,仅供参考。报告内容不能替代线下的面对面诊断与专业医师建议,如有任何疑问或病情变化,请及时到正规医疗机构或专科门诊就诊。
人类医生最终诊断
丛状神经纤维瘤